persian
persian") English (UK)
English (UK) 
![]()
اصطلاح شناسی
شبیه ساز: فراهم کننده ی بستری برای شبیه سازی سیستم های مختلف.
اتمی/ مولکولی: توانایی نرم افزار در شبیه سازی سیستم های اتمی و مولکولی (پیوندها).
بزرگ مقیاس: توانایی شبیه سازی سیستم های پر ذره.
به شدت موازی: توانایی نرم افزار در موازی سازی شبیه سازی های سنگین.
این نرم افزار با در اختیار داشتن میدان نیروهای (Force Field) متنوع فراهم کننده ی بستری مناسب برای شبیه سازی نمونه های مختلف از سیستم های اتمی و مولکولی گرفته تا انواع پروتئین ها و سیستم های زیستی می باشد. از مهمترین ویژگی های این نرم افزار می توان به توان بالای آن در شبیه سازی سیستم های پر ذره اشاره کرد. این بسته ی محاسباتی بر اساس نظریه ی دینامیک مولکولی کار می کند که در ابتدا در فیزیک نظری در دهه 1950 استفاده شد اما امروزه با گسترش حیطه کاربرد آن در علم مواد و زیست مولکولی نیز بکار می رود و قابلیت شبیه سازی سیستم های زیستی و مهندسی را دارا می باشد.
در مورد ساختارهای شبیه سازی شده با استفاده از این نرم افزار می توان به موارد زیر اشاره نمود:
- سیستم های پریودیک (کریستال ها)
- ذرات درشت دانه
- مولکول های ارگانیک
- فلزات
- پلیمرها
- DNA
- پروتئین
- ذرات کروی و بیضی گون با اندازه ی معین
- ...
ویژگی های فنی نرم افزار:
زبان برنامه نویسی: C++
سیستم عامل اجرایی: بر روی تمام سیستم عامل های موجود در بازار قابل اجرا می باشد و تنها کافیست کدها توسط کامپایلر های مخصوص خود اجرا گردند.
لایسنس: تحت پروانه GPL که از زیر مجموعه پروانه های فرهنگ گنو/لینوکس منتشر شده است به این معنی که دسترسی به کدها رایگان بوده و امکان تغییر کدها و انتشار دوباره آن مجاز و قانونی می باشد.
نقاط مثبت نرم افزار:
1- قابلیت اجرا به صورت موازی و سریال (روش معمولی).
2- قابلیت اجرا بر روی GPU ها: GPU به معنی هسته پردازش گرافیکی سیستم می باشد و انجام محاسبات گرافیکی بر عهده ی آن می باشد که این قابلیت موجب کم شدن بار محاسباتی CPU شده و محاسبات سریع تر صورت می پذیرد.
3- اجرای شبیه سازی ها با فایل های Input: این قابلیت به گونه ای موجب کارآمدی و انعطاف نرم افزار گردیده است.
4- متن باز.
5- قابلیت اجرای چند شبیه سازی به وسیله ی یک فایل Input.
6- قابلیت توسعه پذیری بالا.
7- امکان جفت شدن با سایر نرم افزارها.
8- پیش بینی حالت های مختلف شبیه سازی و پختگی کتابخانه های ویژه.
9- مستندات آموزشی و توسعه نرم افزاری کامل و به روز.
10- کاربرد پذیری بالا و خروجی های قابل قبول در جایگاههای مختلف.
نقاط منفی نرم افزار:
1- نداشتن واسط کاربری گرافیکی: برای اجرای نرم افزار نیاز به کار در محیط های متنی و دستورهای متنی داریم.
2- ناتوانی در تولید تصاویر گرافیکی متحرک در شبیه سازی های دینامیک مولکولی: نرم افزار لمپس در حقیقت فقط فایل های مختصات و اطلاعات خروجی را تولید کرده و برای ایجاد تصاویر گرافیکی متحرک به نرم افزارهای کمکی از قبیل VMD نیاز داریم که در ادامه این نرم افزار معرفی و توضیح داده خواهد شد.
3- ناتوانی در تولید نمودارهای اطلاعات خروجی.
4- ناتوانی در تعیین خودکار نیروهای اتمی و مولکولی: در شبیه سازی ها نیاز است که نیروها توسط کاربر تعیین شوند. البته این مسئله به نوعی موجب انعطاف در شبیه سازی شده و موجب آزاد گذاشتن شبیه ساز در انتخاب نیروها و بررسی اثر گذاری خاص آن نیرو شده است.
نمونه های زیر با نرم افزارهای Visualation به حالت گرافیکی در آمده اند:
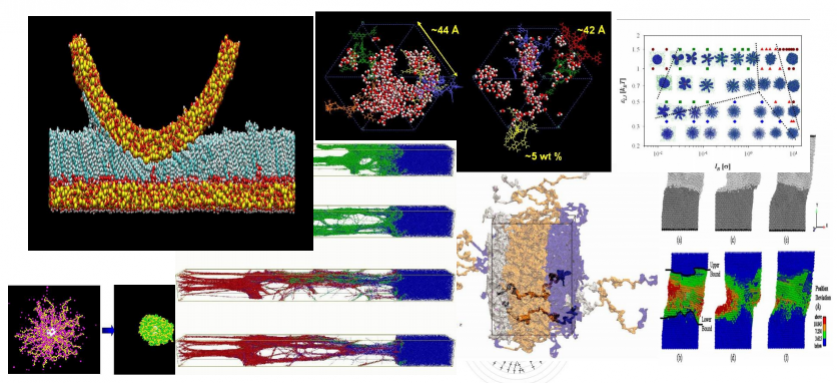
خروجی های نرم افزار:
با استفاده از شبیه سازی های انجام شده توسط این بسته ی محاسباتی می توان بسیاری از ویژگی های ترمودینامیکی و کلاسیکی سیستم های مورد بررسی را بدست آورد. تعدادی از مهمترین خروجی های حاصل از اجرای بسته ی محاسباتی لمپس که با استفاده از آن می توان خواص مکانیکی و ترمودینامیکی مختلفی از سیستم شبیه سازی شده را محاسبه نمود را در زیر فهرست کرده ایم:
1- Log file شامل اطلاعات ترمودینامیکی سیستم شبیه سازی شده.
2- خروجی از مکان و سرعت هریک از اتم های شبیه سازی شده در هر لحظه دلخواه از زمان شبیه سازی شده.
3- نمایش مقادیر انرژی، دما، فشار و ... سیستم شبیه سازی شده.
4- میانگین گیری فضایی و زمانی از مشخصات تک تک اتم ها.
5- ارائه خروجی قابل نمایش گرافیکی از رفتار سیستم در بازه های زمانی شبیه سازی در فرمت هایی مختلف از قبیل XYZ, XTC, DCD, CFG.
و ...
قواعد کلی کدنویسی لمپس
چند نکته اساسی در مورد کلیه کدهای لمپس:
- هر خط غیر خالی به عنوان یک خط کد با معنا توسط لمپس شناخته می شود.
- دستورهای لمپس Case Sensitive هستند، بدین معنی که به بزرگ و کوچکی حروف حساس اند.
- حروف بزرگ (Upper-Case) برای نوشتن نام ها و آی دی ها استفاده می شوند.
- خط هایی از کد که با کاراکتر # شروع می شوند به عنوان توضیحات فرض می شوند و اجرا نخواهند شد و فقط به شبیه ساز در مورد کدها کمک می کنند.
ساختمان کدهای ورودی:
کدهای ورودی لمپس به صورت عمومی(پیشفرض) دارای 4 قسمت است:
1- قالب بندی (Initialization)
2- معرفی اتم ها و مولکول ها (Atom Definition)
3- تنظیمات و پیکربندی ها (Settings)
4- کدهای اجرایی (Run a Simulation)
دو بخش پایانی می توانند در یک کد به طور مکرر تکرار شوند. یعنی می توان با پیکربندی خاصی یک بار کد را اجرا نمود و سپس در ادامه ی کد پس از اعمال تغییرات موردنظر دوباره کدها را اجرا کرد و این روند می تواند به طور متوالی تکرار شده و نتایج خروجی با هم مقایسه شوند. در ادامه به طور خلاصه به تشریح هر کدام از 4 بخش فوق خواهیم پرداخت.
قالب بندی:
در این بخش پارامترهایی تنظیم خواهد شد که نیاز است قبل از تعریف اتم ها و مولکول ها یا خواندن فایل های ورودی، برای سیستم تعریف شده باشند. مهمترین دستورهای مرتبط با این بخش عبارتند از:
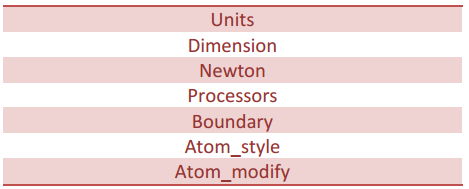
معرفی اتم ها و مولکول ها:
برای معرفی اتم ها و مولکول ها در لمپس 3 روش در دسترس است:
1- خواندن اطلاعات ورودی از فایل های دیتا و ریستارت با دستورهای read_data و read_restart. در این فایل ها می توان ترکیب های مولکولی و ساختارهای مختلف را در شبیه سازی ها وارد نمود.
2- تعریف مستقیم اتم ها و شبکه های اتمی (بدون پیوندهای اتمی پیشفرض) با استفاده از دستورهای create_atom, create_box, region, lattice.
3- ترکیبی از دو روش بالا برای شبیه سازی های بزرگ و پیچیده (به کمک تکرار دستورهای دو شیوه ی فوق).
تنظیمات و پیکربندی ها:
پس از تعریف اتم ها و ساختارهای مولکولی حال سیستم آماده است تا پیکربندی های مختلفی بر آن اعمال شود. مانند حوزه و ضریب نیروهای بین مولکولی، پارامترهای اجرایی شبیه سازی، قواعد خروجی های شبیه سازی و ... . در ادامه به بخشی از تنظیمات قابل تعریف برای سیستم های شبیه سازی اشاره می کنیم:
- ضریب و حوزه تاثیر نیروهای بین اتمی و مولکولی با دستورهای زیر قابل تنظیم اند.
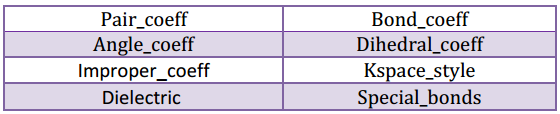
- پارامترهای مختلف شبیه سازی با دستورهای زیر تنظیم می شوند:
استفاده از دستورهای fix برای اعمال کردن ویژگی های مختلف اعم از حالت ها و شرایط مرزی، یکپارچگی زمان و ... .
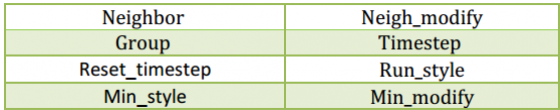
- ویژگی های خروجی نیز با دستورهای زیر قابل تنظیم هستند:

کدهای اجرایی:
شبیه سازی مولکولی با استفاده از دستور run در لمپس اجرا می شود و دیگر کدهای مربوط به اجرای موازی(Parallel) در این بخش قرار می گیرند.
- دستورهای ذکر شده ی بالا تنها بخش کوچکی از کل دستورهای قابل اعمال و اجرا در لمپس هستند به همین دلیل نیاز است که برای آشنا شدن کامل با دستورهای لمپس به Manual این نرم افزار رجوع شود.
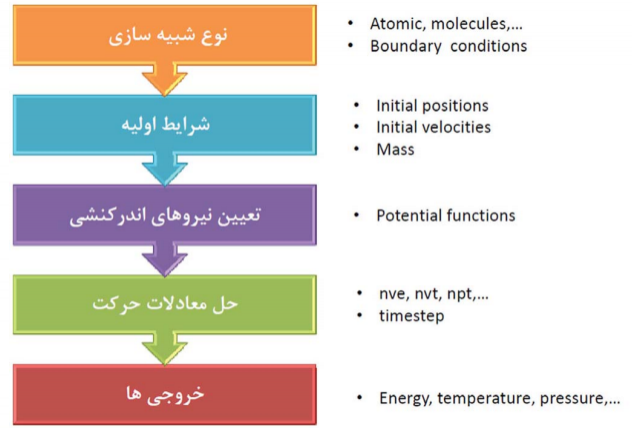
نحوه نوشتن یک فایل ورودی:
در حالت کلی input file ورودی در لمپس باید دارای بخش های کلی زیر باشد:
تحلیل نمونه کد شبیه سازی دینامیک مولکولی در لمپس:
قالب بندی:
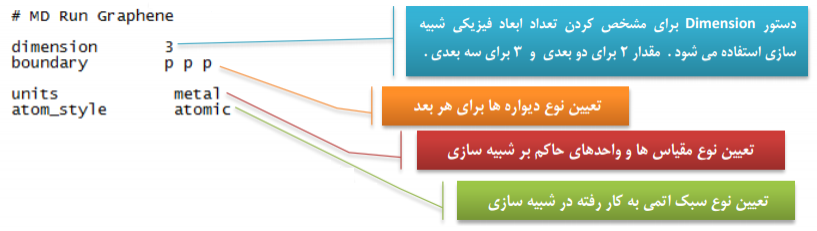
معرفی اتم ها و مولکول ها:
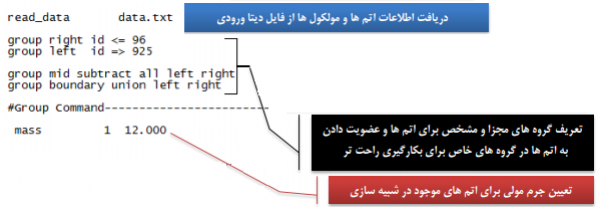
تنظیمات و پیکربندی ها:

نمایش خروجی ها:
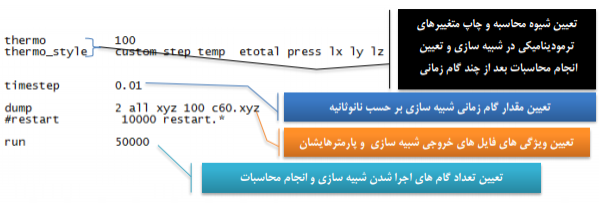
----------------------------------------------
منبع : lammps.ir