persian
persian") English (UK)
English (UK) 
در این آموزش به بررسی خواص ساختاری جامد سیلیکون پرداخته خواهد شد. قبل از شروع محاسبه ابتدا مقداری در مورد تئوری های لازم صحبت می شود.
اول از همه باید در نظر داشته باشیم که راحت ترین پتانسیل ترمودینامیکی در آنالیز نظری ابتدا به ساکن انرژی کل در دمای صفر کلوین است که تابع کمیت زیر است:
N تعداد و گونه های اتمی حاضر در سلول واحد
Ω حجم سیستم که به بردار های شبکه و ثوابت شبکه بستگی دارد
S که در دمای صفر مقدار آن صفر است
خیلی ساده است که ما محاسبات ساختار الکترونی را در حجم ثابت انجام بدهیم.
اولین محک: تعیین پیش بینی نظری Ω0 و B برای ساختار های بلوری شناخته شده
ابتدا باید یک سری کمیات را تعریف کنیم:
انرژی 
فشار 
مدول حجمی 
سوالی که پیش می آید این است که چرا ما دنبال Ω0 و B هستیم؟ به دو دلیل:
- می توانیم آنها را با دقت زیادی اندازه گیری کنیم
- در دمای صفر مطلق میشود آنها را برون یابی کرد
در جدول زیر که از کتاب کیتل برداشته شده پایدارترین شکل بلوری عناصر در دمای اتاق لیست شده است.
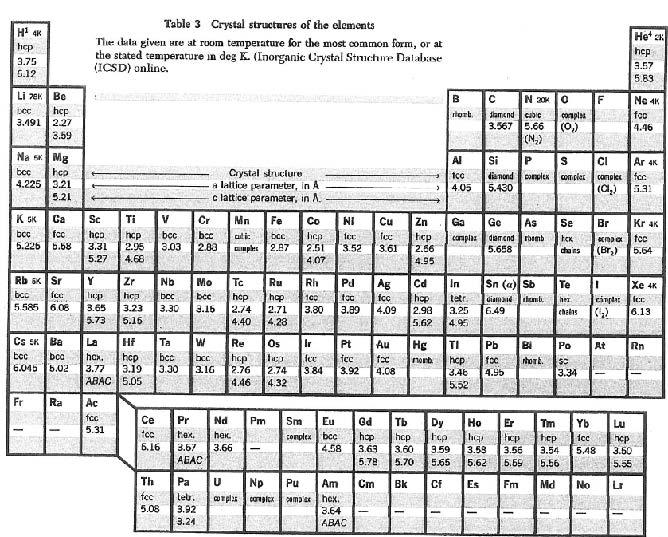
همچنین در جدول زیر که باز هم از کتاب کیتل برداشته شده مدول حجمی و تراکم پذیری لیست شده است.
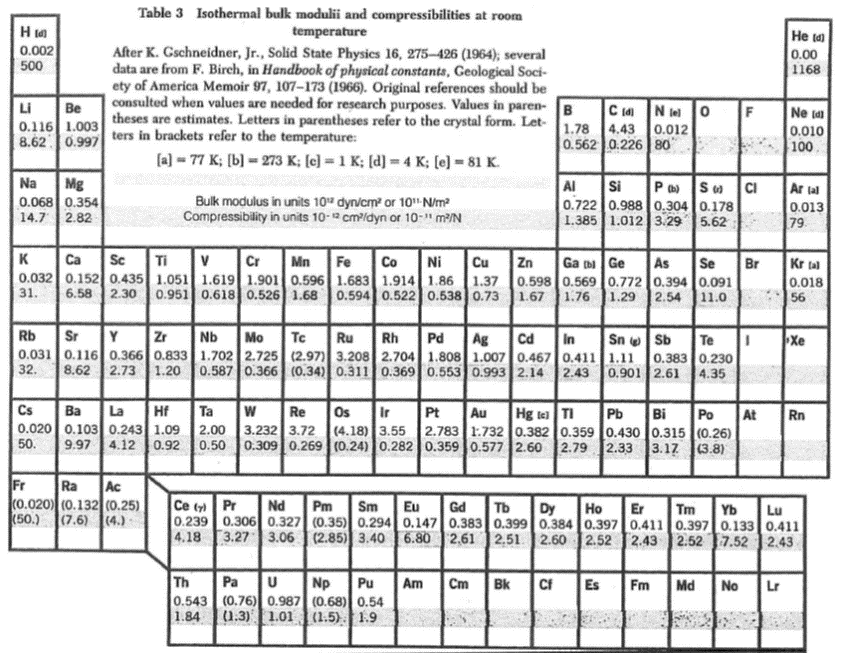
همانطور که میبینید این کمیات با دقت خوبی از طریق تجربه بدست می آیند و میشود با یک برون یابی مقدار آنها در صفر مطلق را پیش بینی کرد پس معیار خوبی برای مقایسه هستند. حال هدف ما این است که این کمیات را از طریق محاسبات ابتدا به ساکن بدست بیاریم.
سیستم مورد بررسی: جامد سیلیکون – جامدی کووالانس که در ساختار الماسی متبلور می شود.
برای شروع فایل های مورد نیاز را از انتهای همین مطلب دانلود کنید و روی سیستم خود در یک جای مشخص از حالت فشرده خارج کنید. بعد از این کار شما یک فایل ورودی Si.fdf خواهید داشت که محتویاتش به شکل زیر است:
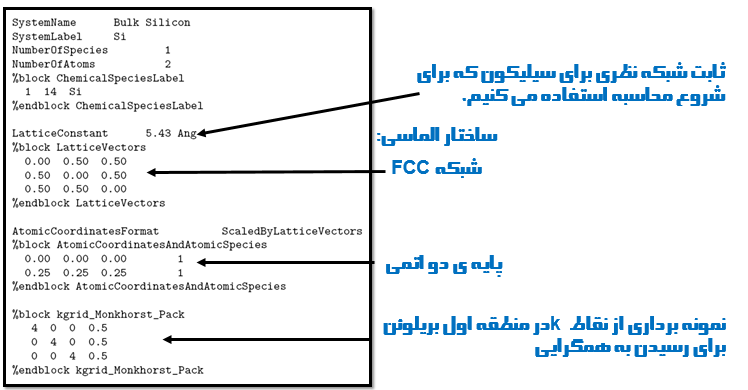
مراحل محاسبه حجم تعادلی و مدول حجمی
مرحله اول: یک ساختار معین را در ورودی تعریف کنید، انرژی E رو برای مقادیر مختلفی از حجم Ω محاسبه کنید (در واقع یعنی هر مرحله حجم رو تغییر بدید که در اینجا چون ساختار مکعبی هست یک ثابت بکه تعریف کردیم و همان را تغییر میدهیم). برای اینکار یه ثابت شبکه تعریف کنید و دستور زیر را در ترمینال بنویسید و اجرا کنید:
siesta < Si.fdf > Si.5.43.out
همانطور که می دانید، اسم فایل خروجی آزاد است و شما هر اسمی می توانید انتخاب کنید ولی در نظر داشته باشید چیزی بنویسید که بعدا که دوباره به این محاسبات مراجعه کردید برای شما معنی داشته باشد. به عنوان مثال در اینجا ما اندازه ثابت شبکه را در اسم فایل خروجی نوشتیم.
در این مثال خاص شما می توانید ثابت شبکه را از مقدار 5.35 آنگستروم تا مقدار 5.49 آنگستروم با گام هایی 0.02 آنگسترومی تغییر بدهید و برای هر مرحله فایل خروجی را ذخیره کنید.
برای محاسبه نمودار انرژی بر حسب حجم شما باید داده های مورد نیاز را استخراج کنید برای این کار می توانید از دستور grep استفاده کنید:
grep "Total =" Si.*.out > Si.evslc.dat
بعد از اجرای این دستور فایل Si.evslc.dat را ادیت کنید و فقط دو ستون به شکل زیر باقی بگذارید. در نظر داشته باشید که در اینجا باید یک تغییر کوچک در این فایل بدهیم که به عنوان ورودی برای یه اسکریپ که بعدا توضیح داده می شود مورد استفاده قرار بگیرد. شما باید یک خط به اول فایل اضافه کنید و نوع شبکه را در آن مشخص کنید (cubic,fcc,bcc,diamond) که در این مثال ما نوع شبکه را diamond در نظر میگیریم.

که ستون اول ثابت شبکه و ستون دوم انرژی کل سلول واحد است. حالا می توانید برای داده ها یه نمودار هم رسم کنید که به شکل زیر می شود
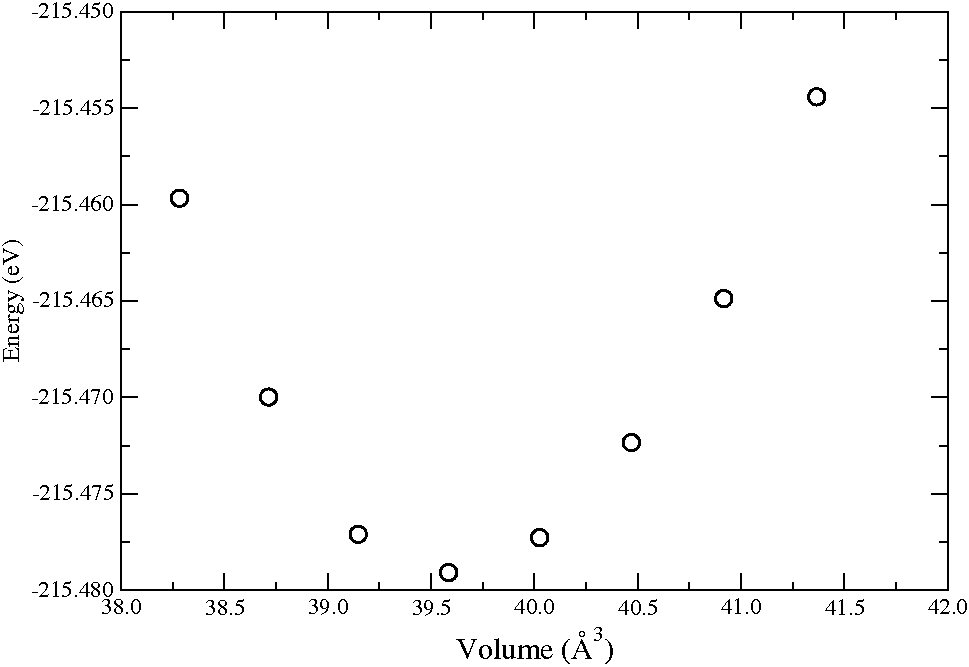
در نظر داشته باشید که در اینجا ما تعداد اتم ها را ثابت نگه داشتیم. همچنین از آنجایی که ساختار اولیه مکعبی بوده و ما اجازه تغییر فقط ثابت شبکه را دادیم پس تقارن را حفظ کردیم همچنین دما صفر است که در اون آنتروپی مقدار صفر دارد.
مرحله دوم: برازش یک تابع تحلیلی مانند معادله حالت مورناگان
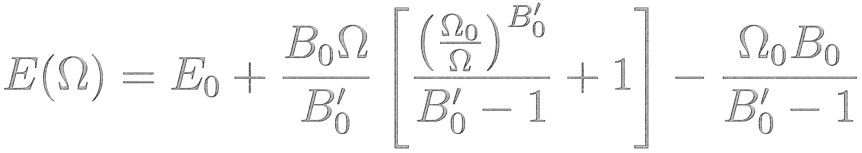
B0 مدول حجمی در حجم تعادلی
B’0 مشتق مدول حجمی بر حسب فشار در حجم تعادلی
E0 انرژی کل در کمینه
برای این کار یک اسکریپت پایتون از قبل نوشته شده و شما می توانید از آن استفاده کنید. برای استفاده از این اسکریپت دستور زیر را وارد کنید:
python fit_results.py Si.evslc.dat
بعد از اجرا این دستور نموداری به شکل زیر به شما نشان داده میشود:
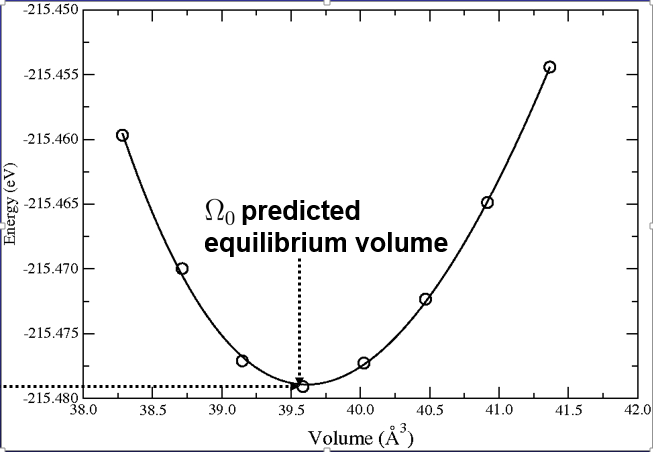
البته مقادیر مدول حجمی و مشتق آن هم در ترمینال برای شما نشان داده میشود.
در ادامه یه مقایسه ای انجام میدهیم بین محاسبات و نتایج تجربی که در جدول زیر مشاهده می کنید:
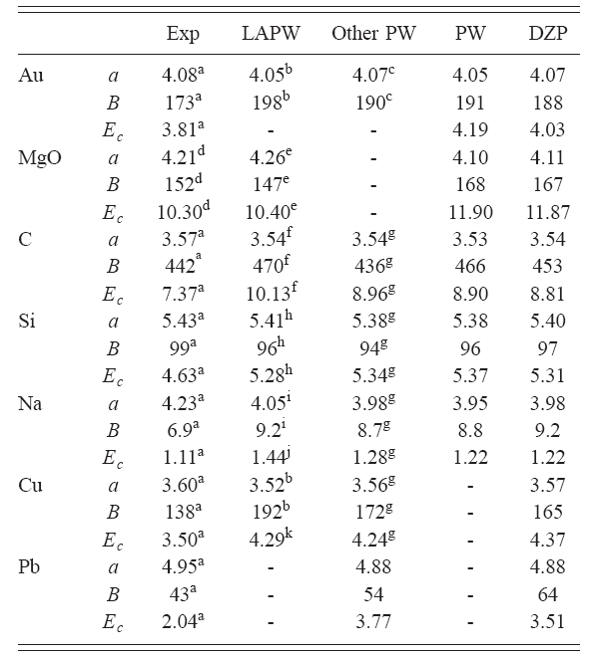
این جدول از مقاله J. Junquera et al., Phys. Rev. B 64, 235111 2001 برداشته شده است.
-------------------------------------------------------------
منبع : comphys.ir